
| 일 | 월 | 화 | 수 | 목 | 금 | 토 |
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | ||||
| 4 | 5 | 6 | 7 | 8 | 9 | 10 |
| 11 | 12 | 13 | 14 | 15 | 16 | 17 |
| 18 | 19 | 20 | 21 | 22 | 23 | 24 |
| 25 | 26 | 27 | 28 | 29 | 30 | 31 |
- scRNAseq
- HTML
- CSS
- Batch effect
- ngs
- cellranger
- javascript
- Git
- python matplotlib
- pandas
- ChIPseq
- Bioinformatics
- 비타민 C
- DataFrame
- julia
- single cell rnaseq
- drug development
- CUT&RUN
- PYTHON
- github
- CUTandRUN
- js
- single cell analysis
- 싱글셀 분석
- scRNAseq analysis
- matplotlib
- drug muggers
- single cell
- EdgeR
- MACS2
- Today
- Total
바이오 대표
[Bioinformatics] NGS 파일 포맷 - fastq, sam, bam, bed, bigwig,,, 본문
[Bioinformatics] NGS 파일 포맷 - fastq, sam, bam, bed, bigwig,,,
바이오 대표 2022. 12. 4. 10:00
NGS 데이터는 sequencer 에 의해서 생성되는 sequencing 파일 (fastq)를 시작으로 모든 분석이 이루어지면서 특정 성격을 띈다. NGS 세계에서 이용되는 많은 데이터 포맷 중, 가장 기본이 되는 파일들의 형식들을 중점으로 정리해보았다.
* sequencers 예시: Illumina HiSeq 2500, Illumina NextSeq 500i, Illumina MiSeq,,,
- FASTQ : sequencing data with scores
- SAM : fastq파일을 aligning (mapping) 한 output 파일 (사람이 읽을 수 있는 버전)
- BAM : SAM 을 binary 한 파일로 사람이 읽을 수는 없지만, 용량을 줄일 수 있다.
- VCF : (Variant Calling Format) 유전자 변이 정보를 담고 있다.
- GTF(GFF3) : (Gene tranfer Format) 유전체 정보를 담고 있어 annotation 할때 사용된다.
- BED : 흔히, 시각화를 위한 basic sequence features 이다. 추후, IGV 와 같은 visualization tool에서 사용될 수 있다.
Fastq Format
Four Lines:
- header @ 에 붙어있는 것은 sequence name/identifier 이고 나머지는 metadata / description이다.
- sequence
- ‘+’ # 분리를 위한 line이다.
- quality score (Phred score) # sequence 와 길이가 같아야 한다.
@60c8e88b-1066 runid=c821e002d29 sampleid=01 read=112044 ch=86 start_time=2019-08-22T08:01:18Z
CGGTAGCGCGTTCAACTCAGGGTGGTGTTTATATGATCGCCGCCTACCGTGACTCAGTGTTGTAGTCATCTATTTTCTGTTGGTACTGATATTGCATGCGATACTTGGGTAGCGAATATCAATAAGCGGAAGATGAAACGAGCGAAGTACGAACGACTACAACAGAATCGAGTC
+
-&+)+)-.0/=+6&'#%(&'$$)#$3%++*%$&&+&%*))+''')&&')*()+-.&&*'.4%&&))(,-.-'==A7*()%'$'*&6300@89:5<,$$.)()%$$$*/1*&%0++15.&4'()$%1602(*.2,#$%$.%&'*$&$$%$%&%&'32:;?:5+/*-*..,%#%''
** header을 글 없는 숫자(free text) 로만 나타내는 경우도 있다
- ex) @EAS139:136:FC706VJ:2:2104:15343:197393 1:Y:18:ATCACG
SAM Format
SAM Header
@ 로 시작되는 모든 Line (@HD: header line, @SH: reference seq dictionary,,, )
11 SAM Columns
- Read ID
- FLAG mapping information 을 2bit로 설명하는 부분이다 (아래 테이블 참고)
- Mapping REF reference name
- Mapping position read의 시작부분 index
- Mapping quality
- CIGAR tag mapping 형태 (M:matching, I:insertion, D:deletion,, =: identical,,,)
- RNEXT mate 의 reference name
- PNEXT mate 의 positon 정보
- TLEN Template length
- SEQ
- QUAL read quality
- TAGS 를 추가로 붙일 수 있다. for extra information
@HD VN:1.6 SO:coordinate
@SQ SN:ref LN:45
r001 99 chr14 7 30 8M2I4M1D3M = 37 39 TTAGATAAAGGATACTG *
r002 0 chr12 9 30 3S6M1P1I4M * 0 0 AAAAGATAAGGATA *
r003 0 * 9 30 5S6M * 0 0 GCCTAAGCTAA * SA:Z:ref,29,-,6H5M,17,0;
r004 0 chr10 16 30 6M14N5M * 0 0 ATAGCTTCAGC *
r003 2064 chrX 29 17 6H5M * 0 0 TAGGC * SA:Z:ref,9,+,5S6M,30,1;
r001 147 chr2 37 30 9M = 7 -39 CAGCGGCAT * NM:i:1
해당 컬럼에 대한 데이터가 없으면 * 로 표시
HEADER 추가 설명
예시에서 볼 수 있는 HD, VN, SO, SN, LN 과 같이 SAM 에 대한 보다 자세한 설명은 https://samtools.github.io/hts-specs/SAMv1.pdf 에서 찾아볼 수 있다.
FLAG 추가 설명

BAM Format
사람이 읽을 수는 SAM 의 binary 형태
VCF Format
VCF Header
## 로 시작되는 모든 Line 은 meta data를 포함한다.
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT SAMPLE … 데이터 컬럼 네임까지 header이다.
VCF Columns: 8 required fields + α
- CHROM 염색체 이름 / 시퀀스 이름
- POS 염색체 위치 / 시퀀스 위치
- ID 해당 변이의 ID ex. dsSNP ID https://www.ncbi.nlm.nih.gov/snp/
- REF reference allele
- ALT alternate allele
- INFO Key-value 로 구성된 data list ( ; 로 구분)
- QUAL alternate allele 관련 Quality (phred-scaled)
- FILTER Pass 면 QC를 통과했다는 뜻 (if not, ref. FILTER in header section)
- FORMAT: sample 추가 필드 목록
- SAMPLEs: format에서 나열한 값을 서술
##fileformat=VCFv4.0
##fileDate=20110705
##reference=1000GenomesPilot-NCBI37
##phasing=partial
##INFO=<ID=NS,Number=1,Type=Integer,Description="Number of Samples With Data">
##INFO=<ID=DP,Number=1,Type=Integer,Description="Total Depth">
##INFO=<ID=AF,Number=.,Type=Float,Description="Allele Frequency">
##INFO=<ID=AA,Number=1,Type=String,Description="Ancestral Allele">
##INFO=<ID=DB,Number=0,Type=Flag,Description="dbSNP membership, build 129">
##INFO=<ID=H2,Number=0,Type=Flag,Description="HapMap2 membership">
##FILTER=<ID=q10,Description="Quality below 10">
##FILTER=<ID=s50,Description="Less than 50% of samples have data">
##FORMAT=<ID=GQ,Number=1,Type=Integer,Description="Genotype Quality">
##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype">
##FORMAT=<ID=DP,Number=1,Type=Integer,Description="Read Depth">
##FORMAT=<ID=HQ,Number=2,Type=Integer,Description="Haplotype Quality">
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT Sample1 Sample2 Sample3
2 4370 rs6057 G A 29 . NS=2;DP=13;AF=0.5;DB;H2 GT:GQ:DP:HQ 0|0:48:1:52,51 1|0:48:8:51,51 1/1:43:5:.,.
2 7330 . T A 3 q10 NS=5;DP=12;AF=0.017 GT:GQ:DP:HQ 0|0:46:3:58,50 0|1:3:5:65,3 0/0:41:3
2 110696 rs6055 A G,T 67 PASS NS=2;DP=10;AF=0.333,0.667;AA=T;DB GT:GQ:DP:HQ 1|2:21:6:23,27 2|1:2:0:18,2 2/2:35:4
2 130237 . T . 47 . NS=2;DP=16;AA=T GT:GQ:DP:HQ 0|0:54:7:56,60 0|0:48:4:56,51 0/0:61:2
2 134567 microsat1 GTCT G,GTACT 50 PASS NS=2;DP=9;AA=G GT:GQ:DP 0/1:35:4 0/2:17:2 1/1:40:3
** INFO, FILTER, FORMAT 관련한 풀이는 header section에서 ##INFO= 혹은 ##FORMAT= 에서 확인 할 수 있다.
흔히 사용 되는 FORMATs:
| GQ | GT(genotype)이 맞을 확률 |
| GT | 0/0 : homozygous 0/1 : heterozygous REF/ALT allele 1/1 : homozygous alternate !! 다시 |
| DP | read depth / filtered depth |
GFT Format
GFT Header
## 로 시작되는 모든 Line 은 meta data를 포함한다.
GFT Columns: 9 required fields (없으면 ‘ . ’로 표시)
- Seq ID
- Source 사용된 algorithm (ex. Genescan) 이나 사용된 프로그램 (ex. Genebank, blast2go)
- Feature type Feature 유형 (ex gene, mRNA, TF, exon, cds, domain, Sequence Ontology nubmer,,, )
- Feature start
- Feature End
- Score E-value (seq. similarity) or p-value (predictions)
- Strand ‘+’ , ‘-’, ‘ . ‘
- Phase Feature 이 cds (coding sequence) 일때, open reading frame 1, 2, 3 를 0, 1, 2 로 표현.
- ( phase 0 이면 첫번째 base부터 codon 시작 , 1 이면 두번째 base 부터 … )
- Attributes Key-value 로 구성된 추가 data list ( ; 로 구분) (ex ID, Name, Alias, Parent …)
GFF 예시: HSP 3 개로 이루어진 alignment
*HSP (high-scoring segment pair) : local alignment with no gaps with highest scores
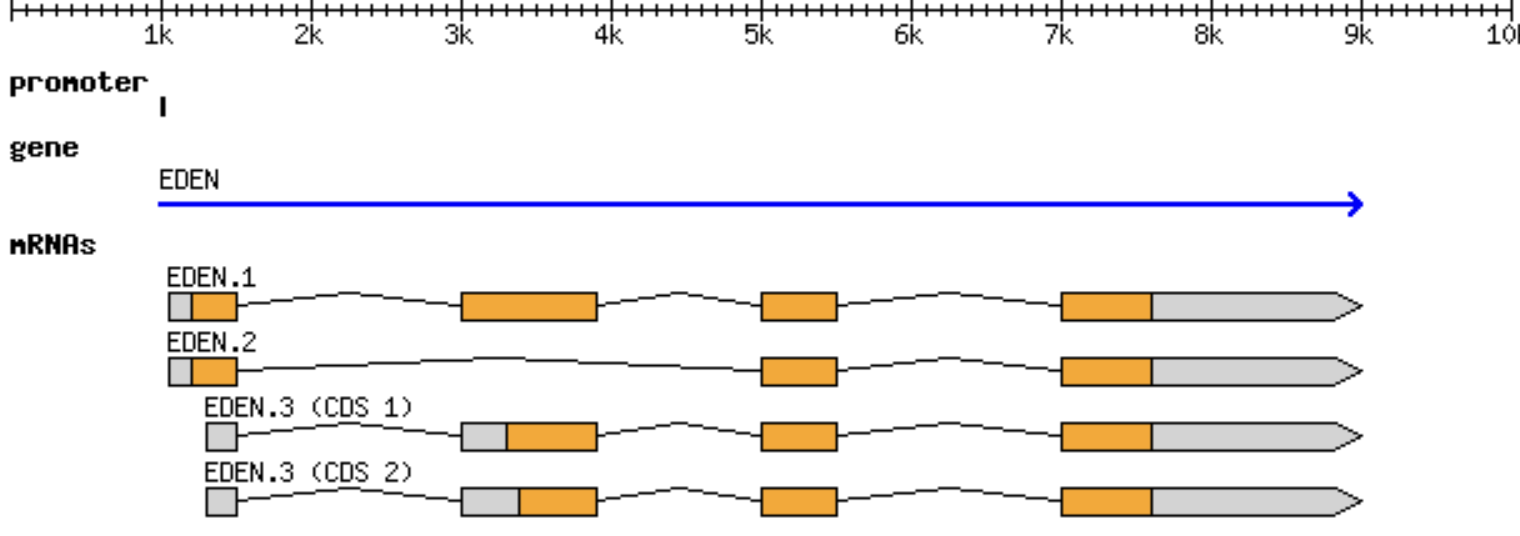
##gff-version 3.1.26
##sequence-region ctg123 1 1497228
ctg123 . gene 1000 9000 . + . ID=gene00001;Name=EDEN
ctg123 . TF_binding_site 1000 1012 . + . Parent=gene00001
ctg123 . mRNA 1050 9000 . + . ID=mRNA00001;Parent=gene00001
ctg123 . mRNA 1050 9000 . + . ID=mRNA00002;Parent=gene00001
ctg123 . mRNA 1300 9000 . + . ID=mRNA00003;Parent=gene00001
ctg123 . exon 1300 1500 . + . Parent=mRNA00003
ctg123 . exon 1050 1500 . + . Parent=mRNA00001,mRNA00002
ctg123 . exon 3000 3902 . + . Parent=mRNA00001,mRNA00003
ctg123 . exon 5000 5500 . + . Parent=mRNA00001,mRNA00002,mRNA00003
ctg123 . exon 7000 9000 . + . Parent=mRNA00001,mRNA00002,mRNA00003
ctg123 . CDS 1201 1500 . + 0 ID=cds00001;Parent=mRNA00001
ctg123 . CDS 3000 3902 . + 0 ID=cds00001;Parent=mRNA00001
ctg123 . CDS 5000 5500 . + 0 ID=cds00001;Parent=mRNA00001
ctg123 . CDS 7000 7600 . + 0 ID=cds00001;Parent=mRNA00001
ctg123 . CDS 1201 1500 . + 0 ID=cds00002;Parent=mRNA00002
ctg123 . CDS 5000 5500 . + 0 ID=cds00002;Parent=mRNA00002
ctg123 . CDS 7000 7600 . + 0 ID=cds00002;Parent=mRNA00002
ctg123 . CDS 3301 3902 . + 0 ID=cds00003;Parent=mRNA00003
ctg123 . CDS 5000 5500 . + 1 ID=cds00003;Parent=mRNA00003
ctg123 . CDS 7000 7600 . + 1 ID=cds00003;Parent=mRNA00003
ctg123 . CDS 3391 3902 . + 0 ID=cds00004;Parent=mRNA00003
ctg123 . CDS 5000 5500 . + 1 ID=cds00004;Parent=mRNA00003
ctg123 . CDS 7000 7600 . + 1 ID=cds00004;Parent=mRNA00003
three spliced transcripts, exons
BED Format
3 required fields (필수)
- chrom name
- chrom start
- chrom end
BED optional fields (추가정보)
- name label
- score study에 따라 다름 (ex ChIPseq - binding affinity, RNA-seq - expression levels, DNA-seq - copy number)
- strand + , -
- thickStart coordination (조정)
- thickEnd
- itemRgb visualization browser (ex. IGV) 에서 보여질 색
- blockCount 해당 sequence 안에서의 block 갯수 (ex. block = exons)
chr7 127471196 127472363 Pos1 0 + 127471196 127472363 255,0,0
chr7 127472363 127473530 Pos2 0 + 127472363 127473530 255,0,0
chr7 127473530 127474697 Pos3 0 + 127473530 127474697 255,0,0
chr7 127474697 127475864 Pos4 0 + 127474697 127475864 255,0,0
chr7 127475864 127477031 Neg1 0 - 127475864 127477031 0,0,255
+ Track Lines (optional)
- Track lines 는 feature lists들의 첫줄에 위치 해야하며, ‘track’ 단어를 시작으로 Key=value pairs
- parameters: name, description, priority, useScore, itemRgb .. etc
track name="ItemRGBDemo" description="Item RGB demonstration" itemRgb="On"
chr7 127471196 127472363 Pos1 0 + 127471196 127472363 255,0,0
chr7 127472363 127473530 Pos2 0 + 127472363 127473530 255,0,0
chr7 127473530 127474697 Pos3 0 + 127473530 127474697 255,0,0
chr7 127474697 127475864 Pos4 0 + 127474697 127475864 255,0,0
'Bioinformatics > NGS 기본지식' 카테고리의 다른 글
| [single cell Analysis] 싱글셀 분석 기본 다지기 4 - 클러스터 visualization (UMAP) (0) | 2023.02.23 |
|---|---|
| [single cell Analysis] 싱글셀 분석 기본 다지기 2 - normalization & batch correction (1) | 2023.02.20 |
| [single cell Analysis] 싱글셀 분석 기본 지식 다지기 1 (0) | 2023.01.30 |

