
| 일 | 월 | 화 | 수 | 목 | 금 | 토 |
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | ||
| 6 | 7 | 8 | 9 | 10 | 11 | 12 |
| 13 | 14 | 15 | 16 | 17 | 18 | 19 |
| 20 | 21 | 22 | 23 | 24 | 25 | 26 |
| 27 | 28 | 29 | 30 |
- Git
- drug development
- PYTHON
- DataFrame
- js
- single cell
- ChIPseq
- matplotlib
- CUTandRUN
- MACS2
- Batch effect
- HTML
- EdgeR
- pandas
- javascript
- 싱글셀 분석
- drug muggers
- github
- python matplotlib
- Bioinformatics
- CUT&RUN
- single cell analysis
- 비타민 C
- scRNAseq analysis
- single cell rnaseq
- CSS
- ngs
- scRNAseq
- cellranger
- julia
- Today
- Total
바이오 대표
[ CITE-seq DE Analysis ] Differential expression analysis for the protein component of CITE-seq data (CiteFuse) 본문
[ CITE-seq DE Analysis ] Differential expression analysis for the protein component of CITE-seq data (CiteFuse)
바이오 대표 2022. 1. 27. 19:34
CITE-seq 은 single cells 에서의 [1] Transcripts [2] Extracellular proteins 의 quantification 을 동시에 구할 수 있다. 기본 scRNA-seq (single cell RNA-seq) 으로는 알아 낼 수 없는 post-transcriptional gene regulation 이나 cell-surface protein levels 와 같은 phenotypic information을 알수 있다.
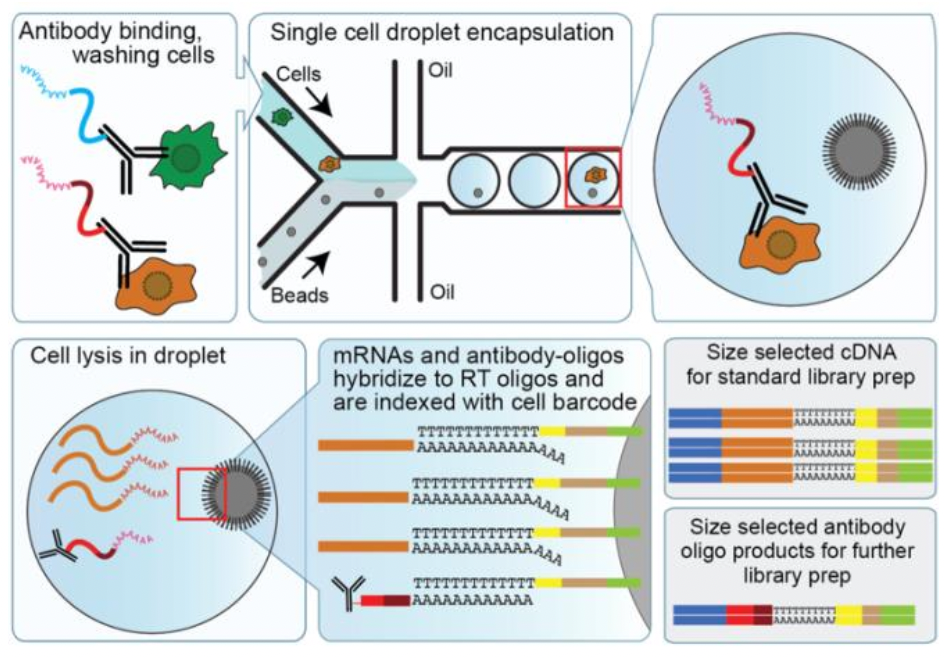
CITE-seq 데이터를 이용해서 DE Analysis 를 진행해보았다. 가장 대표적인 R Bioconductor package CiteFuse 를 이용하였다. CiteFuse 는 transcript data와 protein data 를 구별하여 다루고 각각의 datatypes 에 Wilcoxon rank sum (WRS) 을 이용하여 DE 한 features 을 찾아낸다. 하나의 취약점이라 하면 WRS 을 이용하기에, 만약 low/zero counts 가 많다면 데이터 유대관계로 인해 분석에 어려움이 생긴다.
# CiteFuse Functions
| preprocessing( ) | 데이터 --> SingleCellExperiment 객체 (assay, ADT, HTO) |
| normaliseExprs( ) | to scale the alternation expression (ex) log-transformation of count) |
| crossSampleDoublets( ) | cross-sample doublets 을 확인해주는 function --> saved as $doubletClassify_between_label , $doubletClassify_between_class |
| plotHTO( ) | HTO count의 pairwise sactter을 보여준다. # co-expression of orthologocal HTOs 가 doublets 으로 고려된다 |
| CiteFuse( ) | Similarity network fusion을 이용해서 matrix들을 융합시켜준다 --> $SNF_W 뿐아니라, $RNA_W 와, $ADT_W도 만들어준다 |
| spectralClustering( , K) | clustering 방법 |
| DEgenes( ) | calculates differentially expressed (DE) genes --> $DE_res_RNA_filter 와 $DE_res_ADT_filter |
| DEcomparisonPlot( ) | gene 과 ADT 의 DE analysis 를 비교하여 보여준다 |
| Gene-ADT network | gene 과 ADT 의 연결성을 보여준다 |
Project
해당 프로젝트는 CITE-seq DE Analysis 를 protein Component 에 초점을 두어 진행하였고 WRS 의 취약점을 보안하고 보다 강한 statistical 분석을 위해 distributional assumptions (Poisson,Negative Binomial) 을 하였다. Clustering 전까지는 CiteFuse vignette 과 같이 진행하였다. https://sydneybiox.github.io/CiteFuse/articles/CiteFuse.html
Packages
library(CiteFuse)
library(scater) # Single-Cell Analysis Toolkit
library(SingleCellExperiment)
library(DT) # 대화형 DataTables
library(MASS)
library(ggplot2)
library(edgeR)Data
data("CITEseq_example", package = "CiteFuse")
names(CITEseq_example) # [1] "RNA" "ADT" "HTO"데이터는 RNA, ADT(Antibody derived tags), HTO(hashtag oligonucleotide count) 3개의 matrices 로 구성되어있다. preprocessing function은 이 3개의 matrices와 common cell names를 이용하여 SingleCellExperiment 객체를 만든다. 해당 객체는 assay, ADT, HTO data 이다.
* ADT 는 cell surface protein 에 직접 붙는 antibody 에 달려있는 barcoding(tag) 이다.
sce_citeseq <- preprocessing(CITEseq_example)
해당 예시는 4개의 samples 에서 데이터를 얻은 것이다.
HTO Normalization and Visualization
normaliseExprs( ) 를 이용해서 log-transformation 과 같은 방법으로 count number 을 조정해 줄 수 있다.
sce_citeseq <- normaliseExprs(sce = sce_citeseq,
altExp_name = "HTO",
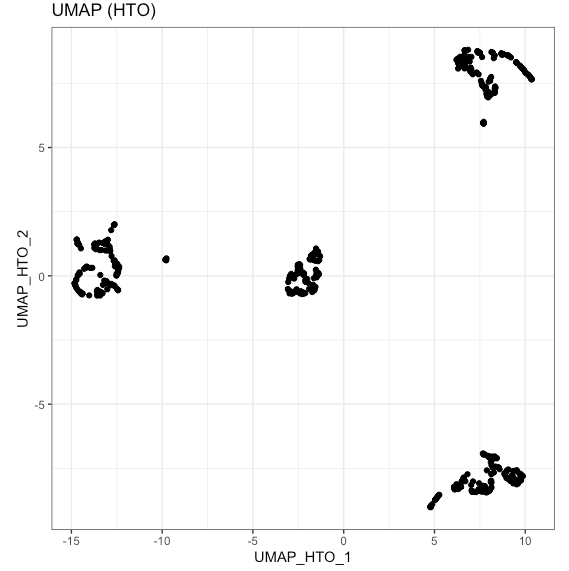
transform = "log")runTSNE or runUMAP 를 이용해서 dimension reduction 도 가능하다. 두 알고리듬의 차이점 참고 클릭
# runTSNE
sce_citeseq <- scater::runTSNE(sce_citeseq,
altexp = "HTO",
name = "TSNE_HTO",
pca = TRUE)
visualiseDim(sce_citeseq,
dimNames = "TSNE_HTO") + labs(title = "tSNE (HTO)")# runUMAP
해당 algorithm 이 compare to TSNE, global structure 을 더 잘 보존한다.
sce_citeseq <- scater::runUMAP(sce_citeseq,
altexp = "HTO",
name = "UMAP_HTO")
visualiseDim(sce_citeseq,
dimNames = "UMAP_HTO") + labs(title = "UMAP (HTO)")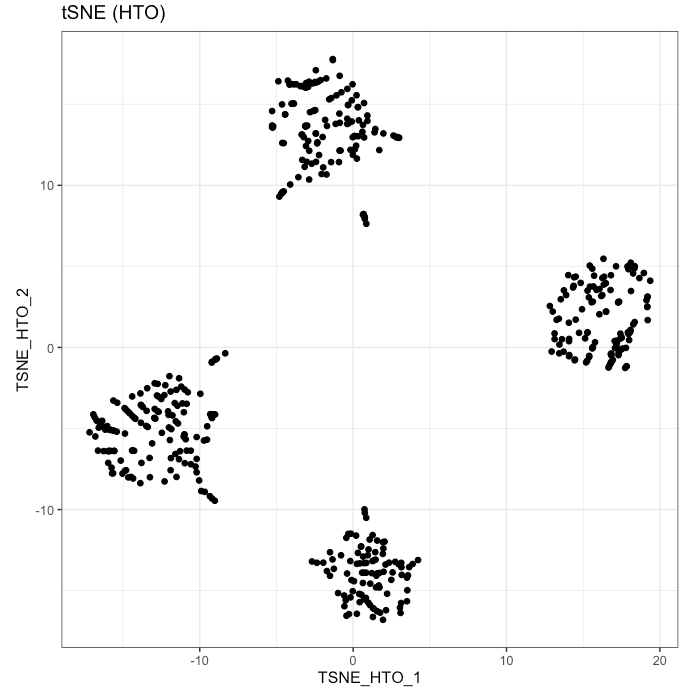
Doublet Identification "cross-sample"
Single cell 데이터를 분석할때, doublet 을 제거하는것은 중요한 step이다. crossSampleDoublets( )를 이용하여 이를 해결할 수 있으면, 해당 function의 output은 'colData' as 'doubletClassify_between_label' 과 'doubletClassify_between_class' 로 저장된다.
sce_citeseq <- crossSampleDoublets(sce_citeseq)

visualiseDim(sce_citeseq,
dimNames = "TSNE_HTO",
colour_by = "doubletClassify_between_label")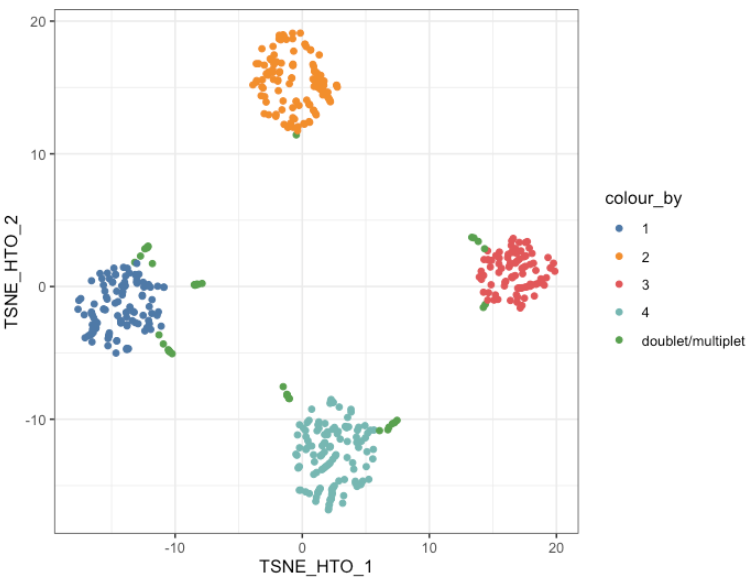
# plotHTO function 을 사용해서도 시각적으로 확인 할 수 있다
plotHTO(sce_citeseq, 1:4)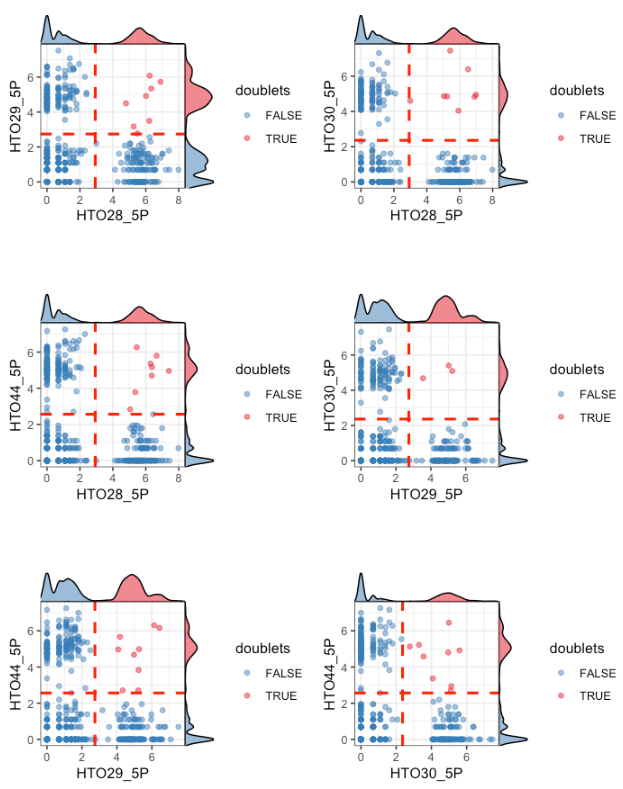
Doublet Identification "within-sample"
within-sample 도 cross-sample 과 같은 방식으로 시각화 할 수 있다.
sce_citeseq <- sce_citeseq[, sce_citeseq$doubletClassify_within_class == "Singlet" & sce_citeseq$doubletClassify_between_class == "Singlet"]Filter out the doublet cells
sce_citeseq <- sce_citeseq[, sce_citeseq$doubletClassify_within_class == "Singlet" & sce_citeseq$doubletClassify_between_class == "Singlet"]
Clustering
먼저, RNA 와 ADT matrices 를 통합시켜줘야한다. 이때 가장 대중적으로 사용되는 SNF (Similarity network fusion)를 이용하였다. https://www.nature.com/articles/nmeth.2810
sce_citeseq <- scater::logNormCounts(sce_citeseq)
sce_citeseq <- normaliseExprs(sce_citeseq, altExp_name = "ADT", transform = "log")
sce_citeseq <- CiteFuse(sce_citeseq)--> sce_citeseq$SNF_W 로 합쳐진 matrix 가 저장된다.
Clustering 방법으로 두가지를 이용하였다.
- Spectral clustering
- Louvain clustering
[1] Spectral Clustering
일단, cluster number 을 구하기 위해 일단 K (clustering 예측 수)로 시작하여 추후, eigen values을 이용해서 clustering 하기 최적의 number 을 구한다.
SNF_W_clust <- spectralClustering(metadata(sce_citeseq)[["SNF_W"]], K = 20)
plot(SNF_W_clust$eigen_values)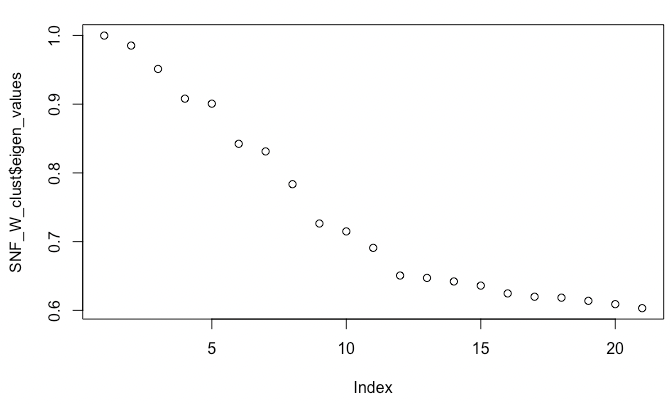
which.max(abs(diff(SNF_W_clust$eigen_values)))
이제 구한 K 값과 spectralClustering( data, K ) 를 이용하여 clustering 을 할 수 있다. CiteFuse 를 사용하면 , SNF_W 뿐 아니라, ADT_W 와 RNA_W 도 생성해줌으로 각각을 비교함으로써 data modality 를 확인할 수 있다.
SNF_W_clust <- spectralClustering(metadata(sce_citeseq)[["SNF_W"]], K = 5)
sce_citeseq$SNF_W_clust <- as.factor(SNF_W_clust$labels)
SNF_W1_clust <- spectralClustering(metadata(sce_citeseq)[["ADT_W"]], K = 5)
sce_citeseq$ADT_clust <- as.factor(SNF_W1_clust$labels)
SNF_W2_clust <- spectralClustering(metadata(sce_citeseq)[["RNA_W"]], K = 5)
sce_citeseq$RNA_clust <- as.factor(SNF_W2_clust$labels)sce_citeseq <- reducedDimSNF(sce_citeseq,
method = "tSNE",
dimNames = "tSNE_joint")
g1 <- visualiseDim(sce_citeseq, dimNames = "tSNE_joint", colour_by = "SNF_W_clust") +
labs(title = "tSNE (SNF clustering)")
g2 <- visualiseDim(sce_citeseq, dimNames = "tSNE_joint", colour_by = "ADT_clust") +
labs(title = "tSNE (ADT clustering)")
g3 <- visualiseDim(sce_citeseq, dimNames = "tSNE_joint", colour_by = "RNA_clust") +
labs(title = "tSNE (RNA clustering)")
library(gridExtra)
grid.arrange(g3, g2, g1, ncol = 2)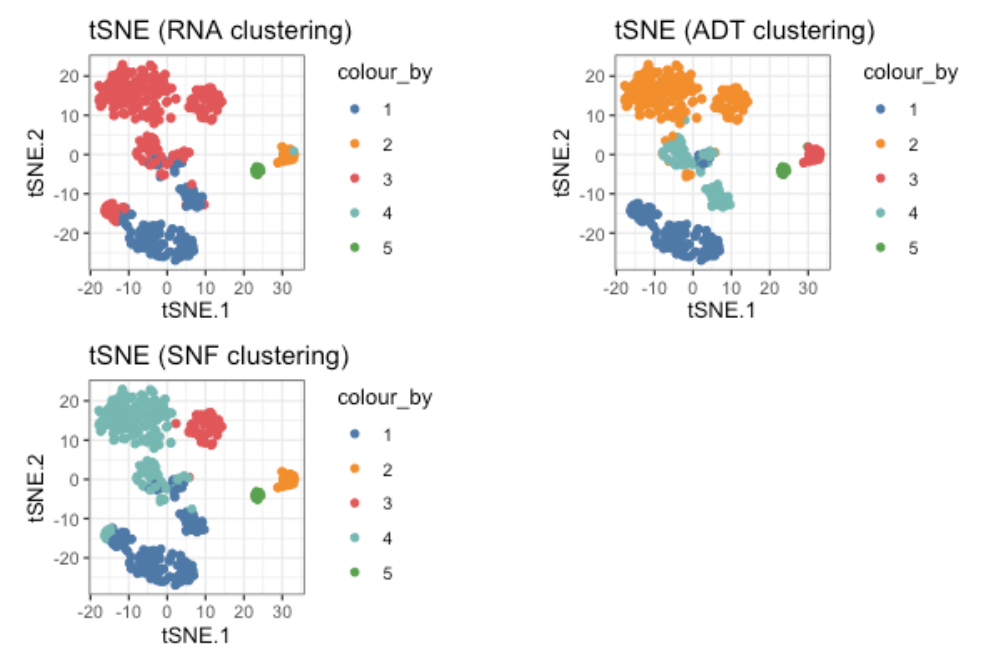
[2] Louvain Clustering
'igraph' package 를 이용하여 louvain clustering 을 할 수 있다.
SNF_W_louvain <- igraphClustering(sce_citeseq, method = "louvain")
table(SNF_W_louvain)
sce_citeseq$SNF_W_louvain <- as.factor(SNF_W_louvain)
visualiseDim(sce_citeseq, dimNames = "tSNE_joint", colour_by = "SNF_W_louvain") +
labs(title = "tSNE (SNF louvain clustering)")
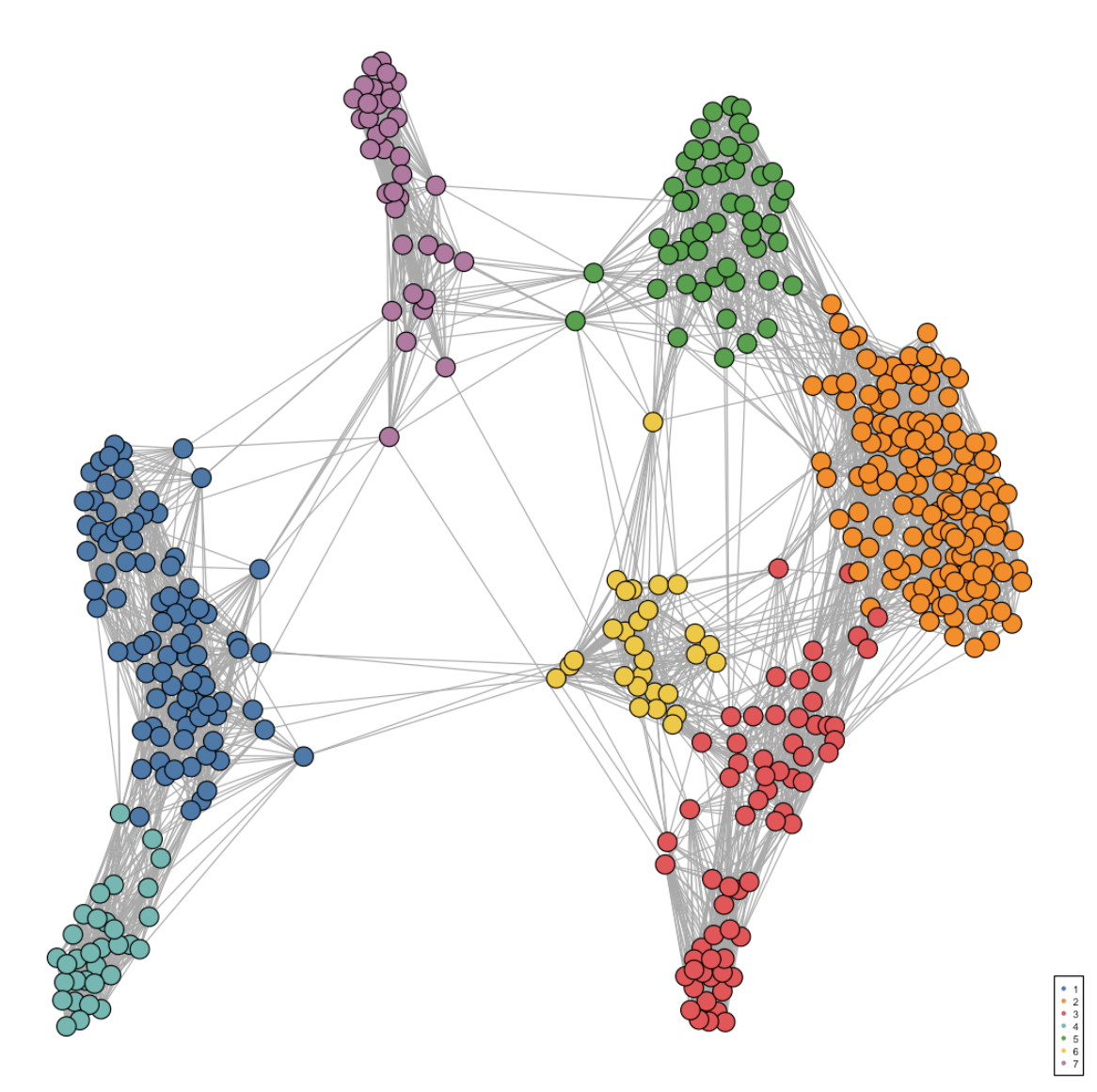
visualiseKNN(sce_citeseq, colour_by = "SNF_W_louvain")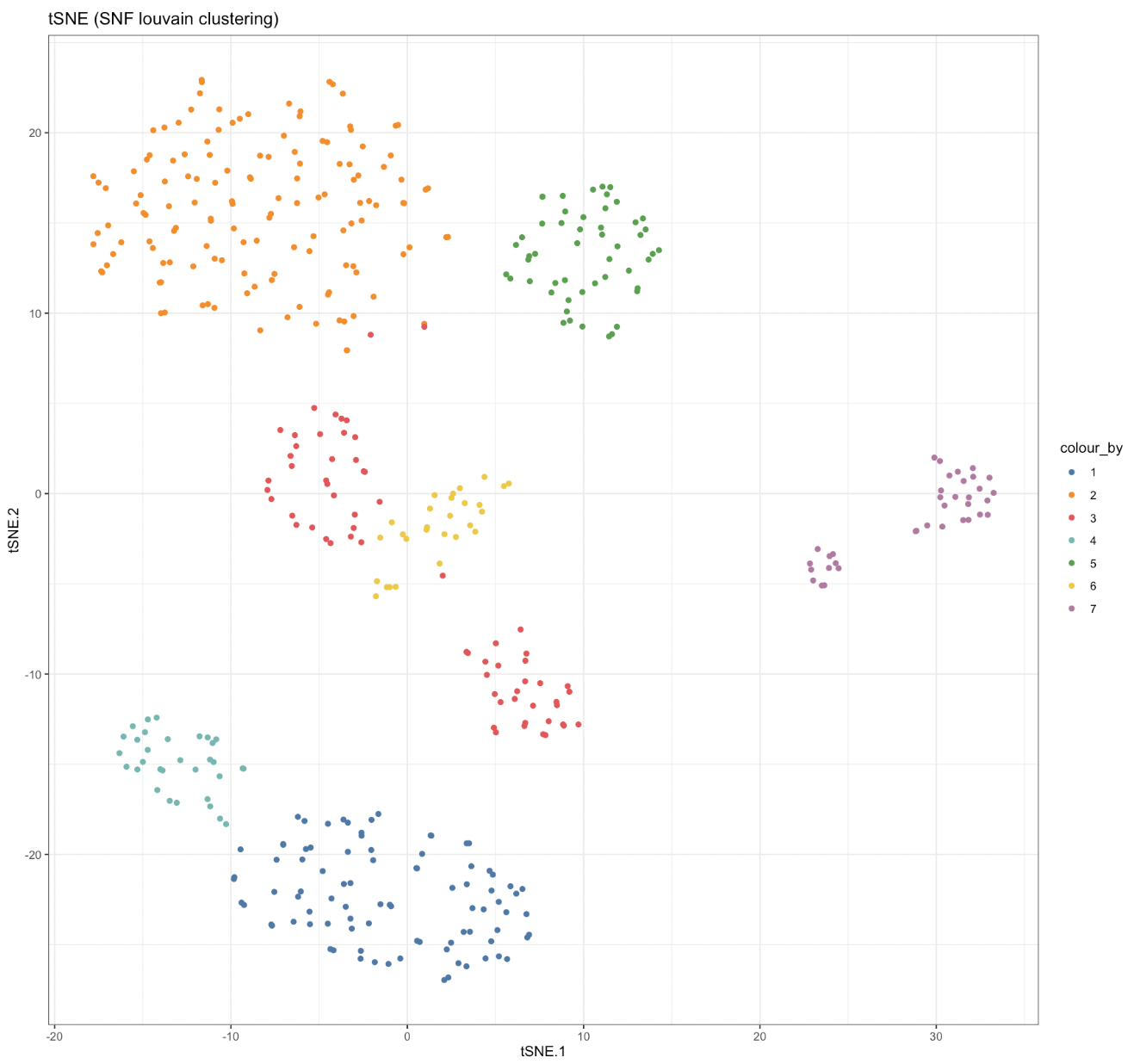
Distribution Assumption
Clustering 후, DE 분석에 앞서 edgeR function 들을 이용하여 distribution Assumptions 을 진행하였다
y <- DGEList(ADT_count)
y <- calcNormFactors(y)
y_filter <- filterByExpr(y, group=group, min.count = 5)--> y$counts 와 y$samples
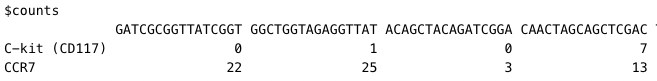

clust.levels = levels(as.factor(group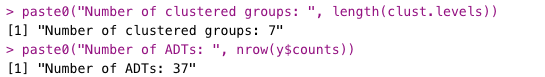
# Poisson Vs NB #
Poisson 과 NB distribution 방식을 비교해 보자면, NB 가 1개의 parameter을 더 갖고있다. 이는, null hypothesis 를 reject 할 수 있는 하나의 옵션이 더 있음을 의미한다. 따라서 NB가 더 나은 성능을 갖고 있을 가능성이 높고 이는 plots 을 통해서도 확인 할 수 있다. 또한 Poisson Distribution 은 mean 과 variance 가 같다고 예측하여 데이터가 평균보다 높을 시 overdispersion 으로 분류하지만 NB 는 좀 더 관대하게 데이터를 다룬다.
37 개의 ADTs 마다 distribution (Poisson vs Negative Binomial)을 예측하는 plot 을 그렸다.두 distribution을 수치로 비교하기 위해, LRT 값과, AIC 값을 이용하였다. 결과적으로 모든 그래프들의 결과들을 통합해보면 LRT 가 낮은 값을 갖고 있는 distribution은 NB AIC 가 Poisson AIC 보다 낮아, 더 나은 성능을 갖고 있음을 보였다.
* LRT (Likelihood ratio test)
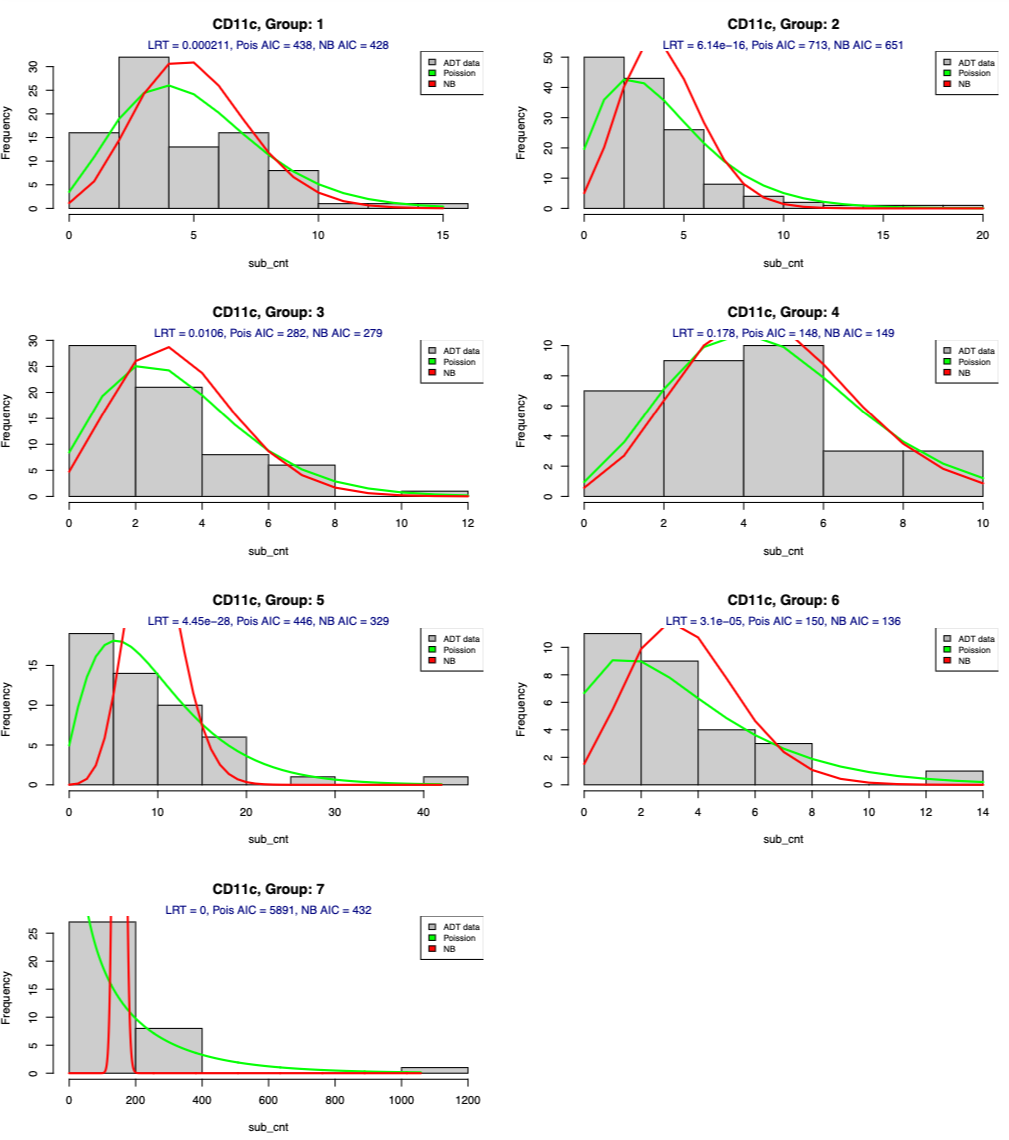
! 아래와 같이 몇몇 그래프는, visually possion distribution 에 더 맞게 보이지만 AIC 수치상으로는 NB이 더 좋은 성능을 보여준다.
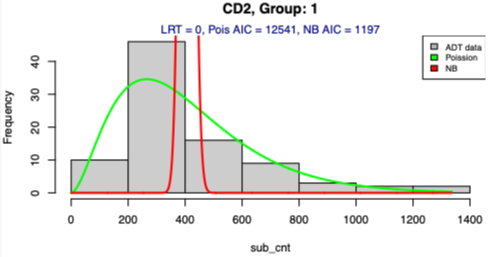
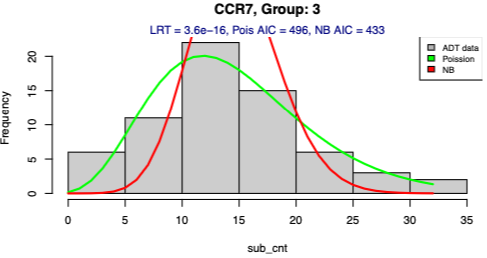
Conclusion:
ADT 데이터를 다루는데에는, NB distribution 을 이용하는 것이 더 알맞다
DE Analysis
분석에 앞서, 데이터들을 빠르고 쉽게 관찰하기위해 시각화를 하는데 visualiseExprs function 을 이용하면 boxplots, violinplots, jitter plots, density plots, and pairwise scatter/density plots 등을 쉽게 구현할수 있다.
Exploration of feature expression
# 예시들
g1 <- visualiseExprs(sce_citeseq,
plot = "boxplot",
group_by = "SNF_W_louvain",
feature_subset = c("hg19_CD2", "hg19_CD4", "hg19_CD8A", "hg19_CD19"))
g2 <- visualiseExprs(sce_citeseq,
plot = "jitter",
group_by = "SNF_W_louvain",
feature_subset = c("hg19_CD2", "hg19_CD4", "hg19_CD8A", "hg19_CD19"))
g3 <- visualiseExprs(sce_citeseq, altExp_name = "ADT",
plot = "density",
group_by = "SNF_W_louvain",
feature_subset = c("CD2", "CD8", "CD4", "CD19"))
g4 <- visualiseExprs(sce_citeseq, altExp_name = "ADT",
plot = "pairwise",
feature_subset = c("CD4", "CD8"))
library(gridExtra)
grid.arrange(g1,g2,g3,g4 ,ncol = 2)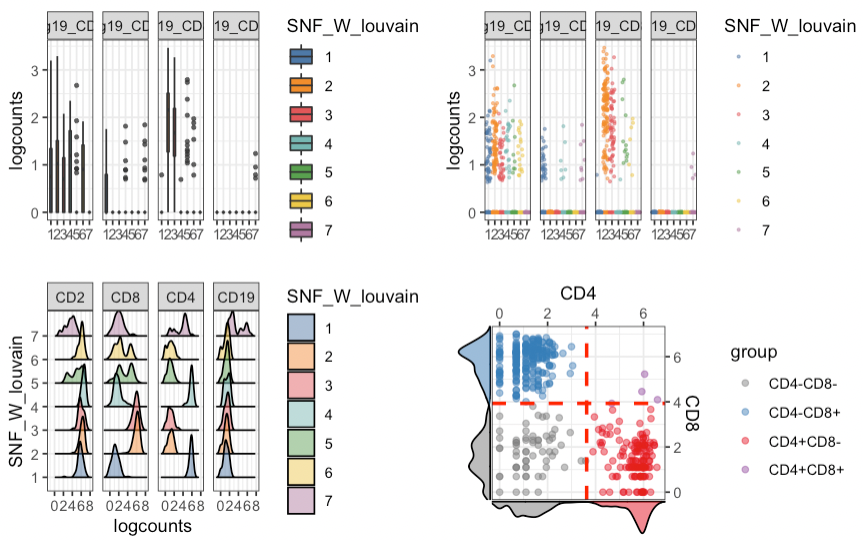
DE Analysis - Wilcoxon Rank Sum 검정
DEgenes( ) 를 이용하여, differentially expressed genes을 계산할 수 있다. 이때, 어떤 group을 분석할 것인지 지정을 해줘야하고 만약 altExp_name = "none" 이면 RNA를 기준으로 DE 가 진행된다.
--> $DE_res_RNA_filter 와 $DE_res_ADT_filter 로 저장이 된다
sce_citeseq <- DEgenes(sce_citeseq,
altExp_name = "none",
group = sce_citeseq$SNF_W_louvain,
return_all = TRUE,
exprs_pct = 0.5)
sce_citeseq <- selectDEgenes(sce_citeseq,
altExp_name = "none")
datatable(format(do.call(rbind, metadata(sce_citeseq)[["DE_res_RNA_filter"]]),
digits = 2))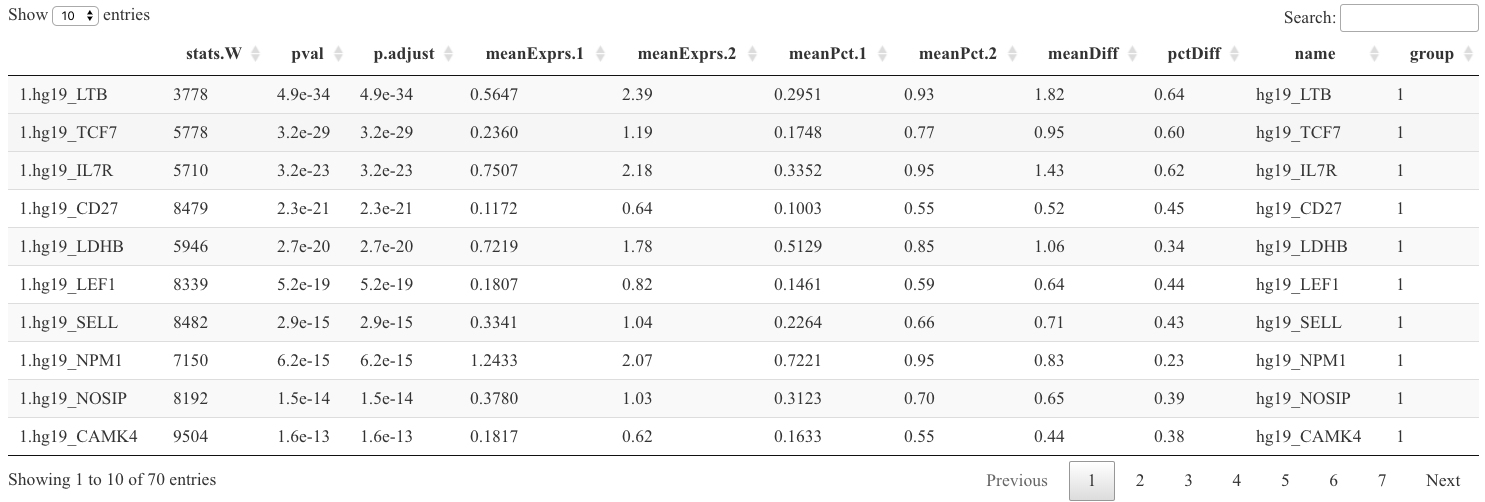
* altExp_name="ADT" 하면 ADT 데이터로 Differential expression analysis 가 진행된다
Visualizing DE Results
DEcomparisonPlot ( )
Gene 과 ADT 의 DE 비교도 가능하다. 아래에서는 관심있는 특정 features 을 직접 입력해주었다.
rna_list <- c("hg19_CD4",
"hg19_CD8A",
"hg19_HLA-DRB1",
"hg19_ITGAX",
"hg19_NCAM1",
"hg19_CD27",
"hg19_CD19")
adt_list <- c("CD4", "CD8", "MHCII (HLA-DR)", "CD11c", "CD56", "CD27", "CD19")
rna_DEgenes_all <- metadata(sce_citeseq)[["DE_res_RNA"]]
adt_DEgenes_all <- metadata(sce_citeseq)[["DE_res_ADT"]]
feature_list <- list(RNA = rna_list, ADT = adt_list)
de_list <- list(RNA = rna_DEgenes_all, ADT = adt_DEgenes_all)
DEcomparisonPlot(de_list = de_list,
feature_list = feature_list)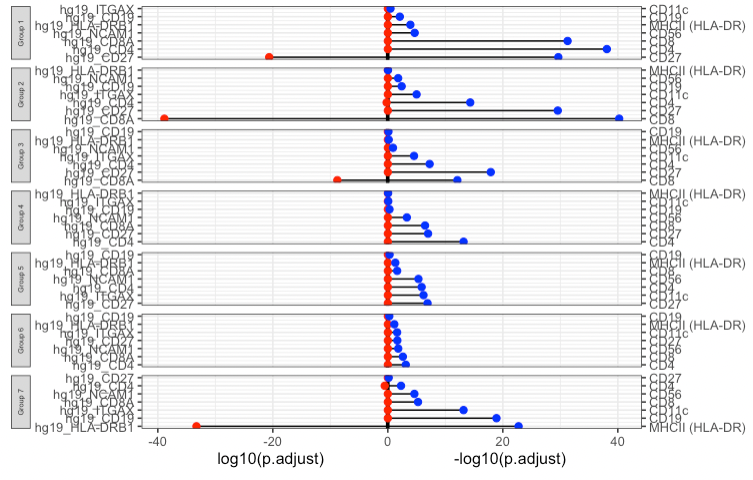
Gene-ADT Network ( )
비교 뿐 아니라 서로의 연관성도 확인 할 수 있다.
RNA_feature_subset <- unique(as.character(unlist(lapply(rna_DEgenes_all, "[[", "name"))))
ADT_feature_subset <- unique(as.character(unlist(lapply(adt_DEgenes_all, "[[", "name"))))
geneADTnetwork(sce_citeseq,
RNA_feature_subset = RNA_feature_subset,
ADT_feature_subset = ADT_feature_subset,
cor_method = "pearson",
network_layout = igraph::layout_with_fr)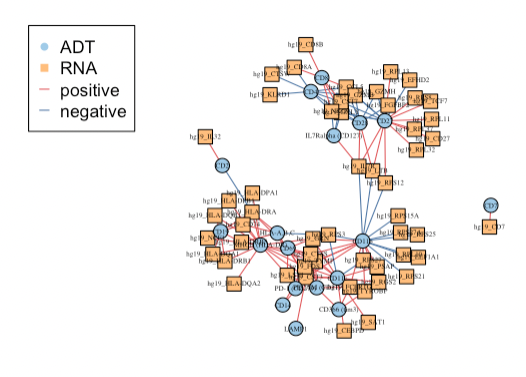
여기에 더해 RNA Ligand-ADT Receptor analysis , Between sample analysis 도 가능하다. 더 궁금하면 웹사이트를 참고하면 좋을 것 같다.


