
| 일 | 월 | 화 | 수 | 목 | 금 | 토 |
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | ||||
| 4 | 5 | 6 | 7 | 8 | 9 | 10 |
| 11 | 12 | 13 | 14 | 15 | 16 | 17 |
| 18 | 19 | 20 | 21 | 22 | 23 | 24 |
| 25 | 26 | 27 | 28 | 29 | 30 | 31 |
- single cell
- drug development
- ChIPseq
- scRNAseq
- CUTandRUN
- github
- matplotlib
- HTML
- cellranger
- Git
- CUT&RUN
- julia
- CSS
- DataFrame
- python matplotlib
- MACS2
- single cell analysis
- PYTHON
- ngs
- EdgeR
- 비타민 C
- scRNAseq analysis
- 싱글셀 분석
- single cell rnaseq
- drug muggers
- pandas
- javascript
- js
- Bioinformatics
- Batch effect
- Today
- Total
바이오 대표
[SEACR 논문] “Peak calling by Sparse Enrichment Analysis for CUT&RUN chromatin profiling” 본문
[SEACR 논문] “Peak calling by Sparse Enrichment Analysis for CUT&RUN chromatin profiling”
바이오 대표 2023. 4. 6. 15:30Background
DNA 의 어느 부분에 protein 이 붙어있는지에 따라, 유전자 발현양이나, 세포의 운명? 이 결정된다. 따라서 해당 위치를 알아내기 위해 사용되는 기술로는 ChiP-seq(protein 과 dna 를 link 하고 dna 자른 뒤 antibody로 추출하는) 과 CUT&RUN(Antibody-directed MNase 가 protein binding 위치를 자르는 기술) 이 있다. ChiP-seq 기술이 좀더 오래 사용되었고, 많은 분석 툴들이 해당 기술에 초점이 맞춰져있어서 이를 CUT&RUN 데이터에 적용시키기 어려운면이 있다. Chip-seq 데이터는 background noise 가 큰 반면, CUT&RUN low read length, low and sparseness background 때문에 ChiP-seq 툴을 사용하면 실제로는 peak 이 아닌데 살짝 시그널이 있다고 positive 라고 예측하는 false-positive가 생긴다. Oversensitivy 하다는 뜻이다. 따라서 SEACR 에서는 specificity 에 초점을 맞춰서 해당 false positive 를 줄이기 위해 global background 를 이용한다.
Sensitivity(= recall) = TP / (TP+FN ) - 실제로 positive 인 것 중 얼마나 맞췄는지
Specificity = TN / (TN + FP) - 실제로 negative 인것 중 얼마나 맞췄는지
Precision = TP / (TP+FP) - positive 라고 예측한 것중 실제 positive 비율
SEACR
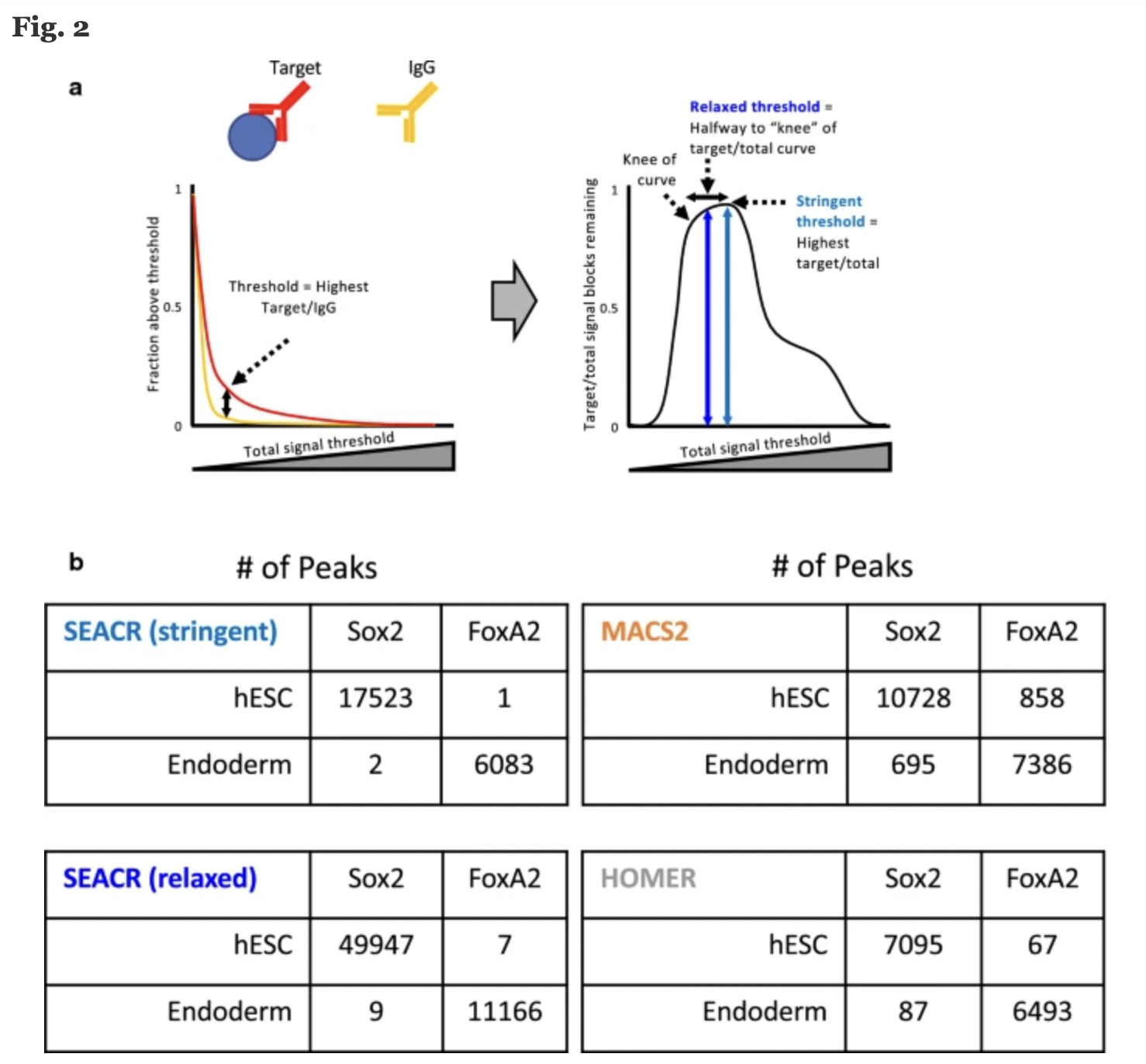
SEACR은 크게보면 global distribution of background signal을 이용해서 threshold 를 정하고 이를 이용하여 peak identity를 하는 알고리즘이라고 보면된다. 좀더 자세히 설명하자면 (Fig.1(a)를 참고바란다), 먼저 CUT&RUN의 target data 와 igG data를 signal block 으로 나눈다. 이때 하나의 signal block은 continuous 하고, nonzero read depth 한 block을 나타내고, 각 block의 signal은 read counts의 합이다. 이러한 signal block들을 이용하여 poportion 그래프를 그릴 수 있는데, 이를 이용해서 calling 되는 target 의 양이 control (igG) 와 가장 차이가 나게되는 threshold를 구할 수 있다. 이 threshold를 넘는 peak들을 calling 한번 해주고, 만약 선정된 peak들중에 igG peak이랑 겹치는 peak (repeated region 혹은 false positve) 또한 제거해준다.

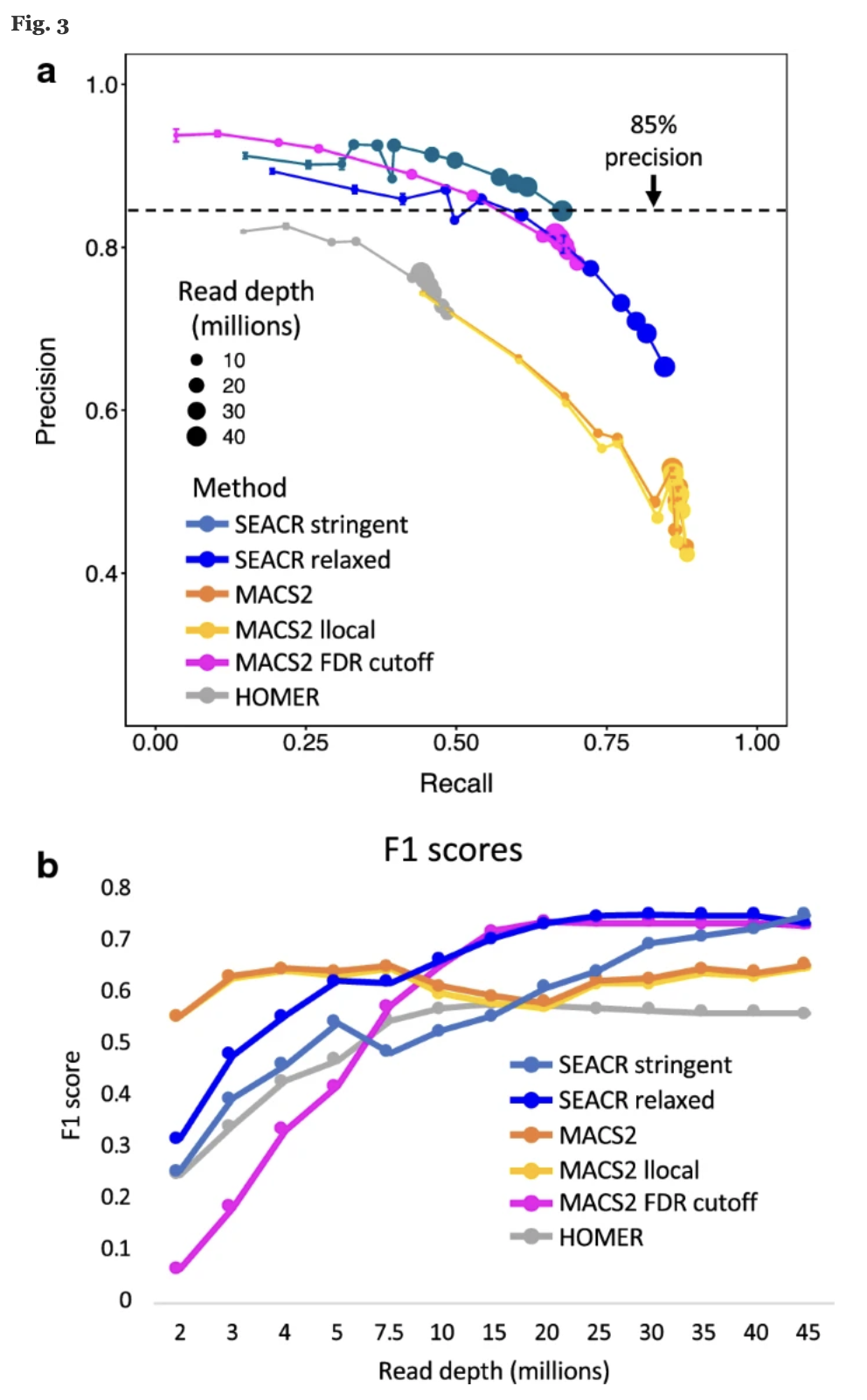
Precision 이 높다는 것은 low false positive rate 을 뜻하고, Recall (sensitivity) 가 높다는 뜻은 low false negative 를 의미한다. F1 score 가 높으면 precision, recall 이 골고루 높다는 뜻이다.
Precision = TP / (TP+FP) - positive 라고 예측한 것중 실제 positive 비율
Sensitivity(= recall) = TP / (TP+FN ) - 실제로 positive 인 것 중 얼마나 맞췄는지
F1 = 2/(1/recall + 1/precision) - harmonic mean of precision and recall
⇒ 결론적으로 SEACR 을 이용하면 false positive 줄일 수 있고 read depth 가 10 million 이상이면 F1 socre 도 제일 좋다는 것을 보여주었다. MACS2 FDR cutoff 가 좋지만, 애초에 ENCODE (gold standard) 데이터가 MACS2 로 추출했다는 것 또 생각해야 할 것 같다.
추가적으로, 해당 논문에서는 SEACR의 peakwidth 가 다른 방법과 비교했을때 가장높고, 사용되는 컴퓨터 파워에서도 (runtime, read memory write memory) 좋은 성능을 보임을 보여준다.
'Bioinformatics > Tools' 카테고리의 다른 글
| [ FRiP 스코어 ] BEDTools, Samtools 이용해서 FRiP 스코어 구하기 (0) | 2023.06.27 |
|---|---|
| [ Peak Annotation] HOMER 이용하여 Peak annotation 하기 - annotatePeaks.pl/loadGenome.pl (0) | 2023.05.16 |
| [ 싱글셀 분석 ] 10x Cell ranger 정복하기 1 (1) | 2023.03.27 |
| [ Cut & Tag / Cut & Run ] Cut & Tag 투토리얼 (0) | 2023.03.18 |
| [ NSG QC / trimming ] TrimGalore (0) | 2023.02.14 |

