
| 일 | 월 | 화 | 수 | 목 | 금 | 토 |
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | ||||
| 4 | 5 | 6 | 7 | 8 | 9 | 10 |
| 11 | 12 | 13 | 14 | 15 | 16 | 17 |
| 18 | 19 | 20 | 21 | 22 | 23 | 24 |
| 25 | 26 | 27 | 28 | 29 | 30 | 31 |
- javascript
- python matplotlib
- 비타민 C
- 싱글셀 분석
- single cell analysis
- PYTHON
- github
- pandas
- single cell rnaseq
- julia
- DataFrame
- CUTandRUN
- MACS2
- Git
- CUT&RUN
- ngs
- drug muggers
- HTML
- CSS
- ChIPseq
- js
- drug development
- scRNAseq
- matplotlib
- Bioinformatics
- scRNAseq analysis
- Batch effect
- EdgeR
- cellranger
- single cell
- Today
- Total
바이오 대표
[NGS scRNAseq] Chromium 10x Illumina의 기본이해(workflow)와 Cell ranger count 본문
[NGS scRNAseq] Chromium 10x Illumina의 기본이해(workflow)와 Cell ranger count
바이오 대표 2022. 12. 18. 16:53
해당 포스트는 Illumina (10x) 기술을 이용한 single cell RNAseq 에 초점이 맞춰져 있습니다.
Chromium 10x single cell - Barcodes and UMI
Paired-end sequencing output 은 주로 5’ → 3’ 방향으로 읽힌 두개의 fastq files 이다. 첫번째 Read 1 (R1) 은 항상 primer 의 Cell barcode + UMI (unique molecular identifiers) 부분을 포함하고 Read 2 (R2)는 reverse sequence를 읽는다(figure 1.3을 참고). Sequencing 으로 얻어낸 reads (containing cell barcode, UMI and cDNA) 를 이용하여, transcipts 의 양을 추정하는 것이 목표이고 여기서 cell barcode 을 이용하여 cell type을 구별하고 UMI 를 이용하여 각 molecule (sequence)를 구별한다.
** Molecules ~ genes


Illumina chromium 10x single-cell sequencing
좀 더 자세한 설명은 http://nextgen.mgh.harvard.edu/IlluminaChemistry.html 참고


Cell Ranger
Cell Ranger은 Chromium single-cell (illumina scRNAseq) 데이터를 mapping 하고, feature(gene)-barcode(cell) matrices를 만들고, clustering 과 다른 분석등을 해주는 분석 파이프라인이다.. RNA reads count matrix 또한 만들어 준다.
즉 upstream analysis, fastq → counts matrix를 만들어주는 소프트웨어 이다.
- Chromium 10x pipeline 알면 좋은 용어들
- GEM well : single 10X chromim chip challel 에서 사용하는 Gelbeads-in-Emulsion이다. 만약 데이터들이 여러 GEM well 에서 생성이 되었다면, GEM 과 다른 GEM 의 bias 를 normalized 해줄 필요가 있다.
- Library (sequencing libraray) : 보통 하나의 GEM well 에서 하나의 10x-barcoded sequencing library를 만들지만 경우에 따라서 여러 libraries을 만들 수 있다.
- Sequencing Run (Flowcell): Sequencing 기계를 한번 돌린것이 one run 이다.
GENERAL WORKFLOW
Chromium 10x pipeline은 다음과 같이 다양한 workflow으로 진행될 수 있다. 만약 여러개의 GEM well 을 이용하였다면 cellranger agrr 를 이용하여 데이터를 합쳐줄 수 있고, 여러 samples 을 하나의 GEM well 을 이용하여 sequencing 을 하였으면, cellranger multi 를 이용할 수 있다. 해당 function들은 여러 데이터를 하나의 큰 matrix로 합쳐, 분석을 좀더 용이하게 할 수 있도록 도와준다.

OUTPUT FILES
Cell ranger 의 output 은 outs/ 폴더에 저장이 된다. 해당 폴더에는 sequencing data, the annotated read sequences, gene expression matrices 등이 존재한다.
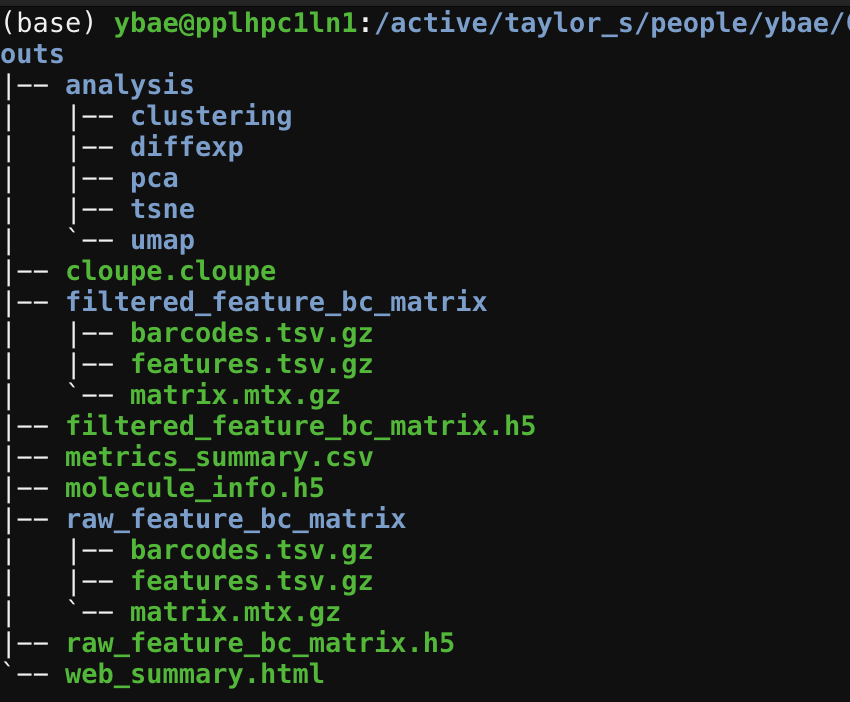
각 파일에 대한 더 자세한 정보
MATRICES
Chromium 10X 는 transcriptomes 뿐아니라, 모든 molecules을 sequencing 하기 때문에 background barcodes 가 생길 수 있다. Cell ranger 는 이를 특정 algorithm 을 사용하여 filtering 해주는데 이는 filtered_feature_bc_matrix 폴더에서 확인 할 수 있다. (위에 output files 참고 바람). Filter이 되지않는 Raw 파일에서 직접 filtering 을 하고싶다면 raw_feature_bc_matrix 폴더를 이용하면 된다. 각각 폴더는 세가지의 파일은 포함하고있다.
- matrix.mtx.gz: reads counts 를 sparse matrices 로 저장한다. ( gene(row) X cell;barcode(column) )
- feature.tsv.gz: 각 row = feature = gene 과 관련된 데이터를 저장한다.
- barcodes.tsv.gz: 각 column = cell 과 관련된 데티어를 저장한다.
WEL SUMMARY .html
cell ranger 가 제공해주는 summary 와 analysis 를 html 형식으로 확인할 수 있다. 다음 figure에서 볼 수 있듯이, 크게 summary 와 gene expression 탭으로 분리가 된다.


해당 요약본에서 주의 깊게 보야할 내용은 다음 포스터에서 확인 할 수 있다.
reference
https://holab-hku.github.io/Fundamental-scRNA/raw2matrix.html#cell-ranger
'Bioinformatics > Tools' 카테고리의 다른 글
| [ Cut & Tag / Cut & Run ] Cut & Tag 투토리얼 (0) | 2023.03.18 |
|---|---|
| [ NSG QC / trimming ] TrimGalore (0) | 2023.02.14 |
| [ 싱글셀 분석 ] cellranger-atac aggr (0) | 2023.02.03 |
| [NGS scATACseq] scATACseq, Cicero를 이용해서 cis-regulatory gene network 분석하기 (0) | 2022.12.28 |
| [NGS scRNAseq] cellranger count 의 output 파일, summary.html 해석 (1) | 2022.12.19 |

