
| 일 | 월 | 화 | 수 | 목 | 금 | 토 |
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | ||
| 6 | 7 | 8 | 9 | 10 | 11 | 12 |
| 13 | 14 | 15 | 16 | 17 | 18 | 19 |
| 20 | 21 | 22 | 23 | 24 | 25 | 26 |
| 27 | 28 | 29 | 30 |
- matplotlib
- github
- drug muggers
- CUT&RUN
- CSS
- MACS2
- pandas
- 싱글셀 분석
- PYTHON
- drug development
- Bioinformatics
- single cell rnaseq
- ngs
- julia
- scRNAseq
- js
- javascript
- HTML
- 비타민 C
- EdgeR
- DataFrame
- CUTandRUN
- single cell
- cellranger
- scRNAseq analysis
- Batch effect
- ChIPseq
- Git
- python matplotlib
- single cell analysis
- Today
- Total
목록전체 글 (217)
바이오 대표
“Current best practices in single-cell RNA-seq analysis: a tutorial” 논문 요약 요약 scRNAseq analysis tools increase → lack of standardization (+ dependency of language) The paper introduces the typical scRNAseq analysis steps as current best-practice recommendations. count matrix pre-processing QC, normalization, data correction, feature selection, dimensionality reduction cell - and gene-level downs..
nextflow 를 이용하여 RNA-seq을 진행하기 위하여 사용하였다. Trim Galore Function: FastQ 파일은 일정하게 QC 하고 trimming 하기 위한 툴. warrper tool of Cutadapt and FastQC + some functions for RRBS type library *Cutadapt: adapter seq, primers, poly-A-tail 등 제거 *FastQC: High throughput sequencing data QC Step 1: Quality Trimming low-quality reads trimmed off using phred quality score Step 2: Adapter Trimming Default: 13pb of Illu..
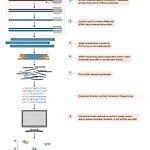 [scRNAseq 논문] 싱글셀 분석 전체 흐름 "A practical guide to scRNAseq for biomedical research and clinical applications"
[scRNAseq 논문] 싱글셀 분석 전체 흐름 "A practical guide to scRNAseq for biomedical research and clinical applications"
"A practical guide to scRNAseq for biomedical research and clinical applications" Abstract Biological sample 의 RNAseq을 이용해서 우리는 mRNA을 발견하고 정략분석을 진행 할 수 있고 이를 이용하여, cellular response를 연구 할 수 있다. 2009년, 첫 scRNAseq 연구가 진행된뒤로, 해당 필드의 많은 발전이 있었다. 해당 논문에서는 scRNAseq 연구를 디자인하기 위한 기본 정보들을 소개한다: hardware, protocol choice, QC, data analysis, biological interpretation. *cellular response: Any process that r..
Cellranger-atac aggr [1] cellranger-atac count [2] cellranger-atac aggr 각각의 GEM well(multiplex data)의 FASTQ 데이터에 cellranger-atac count 를 이용해서 count 를 하고, cellranger-atac aggr를 이용해서 a single peak-barcode matrix 로 합쳐준다. 이때, cellranger에게 aggregation csv 를 이용해 데이터를 주어야 한다. 어떠한 GEM well에 온것인지는 barcode-1, barcode-2 등으로 GEM well suffix(숫자)로 구별할 수 있고 해당 숫자는 Aggregation csv 와 일치한다. Aggregation csv 예시) 1 ..
 [Bioinformatics 논문] TIN score 이용하여 RNA degradation 계산 “Measure transcript integrity using RNA-seq data”
[Bioinformatics 논문] TIN score 이용하여 RNA degradation 계산 “Measure transcript integrity using RNA-seq data”
“Measure transcript integrity using RNA-seq data” Feb.2016 Abstract RNAs 데이터는 보통 archived (저장되어있던) clinical tissue 에서 추출하는데 꽤 degraded 한모습을 보인다. RNA degradation은 RNA-seq read coverage를 왜곡하기도 하고, whole-genome gene expression profiling 에 심각한 영향을 끼치기도 한다. Result TIN (transcript integrity number) 을 이용해서 RNA degradtion 을 measure 할 수 있다. Calibrating gene expression counts with TIN scores 를 통해서 by fals..
Introduction to single-cell RNA-seq Human tissue 에는 엄청나게 다양한 cell types, states, interactions들이 있다. 해당 tissues와 celltypes을 좀 더 자세히 이해하기 위해 single cell RNAseq를 이용할 수 있고 이를 이용해 individual cells에서의 gene express를 확인 할 수있다. 사용예시 하나의 tissue 에 존재하는 cell types 알아낼 수있다. 알려지지 않거나 드문 cell types / states를 알아낼 수 있다. Differentiation process 나 시간/state 의 흐름에 따른 gene expression 변화를 알 수 있다. condition 에 따른 특정 cel..